
图1 6号残留溶剂气相色谱图
张青龄
(福建省粮油质量监测所,福建 福州 350002)
摘 要:应用气相色谱仪结合气相色谱质谱联用仪分析测定大豆原油中残留6号溶剂的组成及各组分的面积百分含量,揭示6号溶剂在各批次的大豆原油中各组成比例差异显著。以新鲜机榨大豆油作基质配制各组分的标准混合溶液,分析计算各组分的相对质量校正因子在0.34~0.81之间,用配对t检验法验证采用各组分标准溶液,以外标法分别定量后加和计算的方法可实现对不同批次样品中溶剂残留量的准确测定。
关键词:大豆原油;6号溶剂残留;组成与比例;相对质量校正因子;配对t检验
6号溶剂是由石油直馏馏分、重整抽余油或凝析油馏分经精制而成的食用油抽提溶剂,该溶剂为混合溶剂,其主要成分为液体烷烃类化合物。食用油中残留的6号溶剂是食用油采取浸出加工过程中残留在油脂中的有毒有害物质,它会麻醉人的中枢神经系统,对人体产生一定的危害[1]。GB 1535—2003[2]大豆油产品标准中限定大豆原油的溶剂残留量为100 mg/kg,浸出的一级和二级大豆油的溶剂残留量为不得检出,三级和四级大豆油的溶剂残留量为50 mg/kg。目前食用油中溶剂残留量的测定国家标准GB 5009.262—2016[3]及多数文献[4-6]中都使用6号溶剂或正己烷为标样,面积归一法定量,但有文献[1,7-8]研究发现不同来源的6号溶剂配制标准溶液时测定结果差异很大。本研究应用气相色谱仪结合气相色谱质谱联用仪分析测定大豆原油中残留6号溶剂的内在组成及各组分在气相检测器上的响应,为准确测定食用油中溶剂残留提供参考依据。
1.1.1 材料
大豆原油(厂家送样);6号溶剂(厂家提供);新鲜机榨大豆油(厂家提供);标准品:2-甲基戊烷、3-甲基戊烷、正己烷、甲基环戊烷、环己烷;正庚烷(色谱纯);N-N二甲基乙酰胺(分析纯)。
1.1.2 仪器
GC 2010 Plus气相色谱仪:附氢火焰离子检测器,日本岛津公司;GCMS-QP 2010 plus气相色谱质谱联用仪:日本岛津公司;PE TurboMatrix 16顶空进样器;20 mL顶空进样瓶。
气相色谱柱为rtx-5弹性石英毛细管柱(30 m×0.25 mm×0.25 μm);汽化温度250 ℃,检测器温度300 ℃,柱箱:初始温度50 ℃,保持3 min后,以1 ℃∕min的升速,至55 ℃,再 以40 ℃∕min升至180 ℃;载气:高纯氮气(气相色谱仪用)或高纯氦气(气相色谱质谱联用仪用),分流比为20∶1,恒线速度模式,柱流量,0.5 mL/min。
顶空进样器炉温60 ℃,取样针90 ℃,传输线120 ℃,保温30 min,加压2 min,拔针0.3 min,进样量100 μL。
质谱接口温度280 ℃,离子源温度200 ℃,电离方式EI源,电离电压70 eV,采集方式:Scan,扫描 m/z 20~300,溶剂延迟时间2.1 min。
称取5.0 g大豆原油样品于20 mL气化瓶中,封盖后放入顶空进样器中备用。
1.4.1 10 mg/mL 6号溶剂标准溶液配制
吸取适量6号溶剂,各加入装有5.0 g新鲜压榨大豆油的20 mL气化瓶中,配制成浓度分别为10、20、50、100、200 mg/kg的标准溶液系列,封盖后放入顶空进样器中。
1.4.2 2-甲基戊烷、3-甲基戊烷、正己烷、甲基环戊烷、环己烷(各10 mg/mL)标准混合溶液配制
吸取适量标准物质,各加入装有5.0 g新鲜压榨大豆油的20 mL气化瓶中,配制成浓度分别为10、20、50、100、200 mg/kg的标准混合溶液系列,封盖后放入顶空进样器中。
样品按1.3处理,用顶空进样器取样,上机进行气相色谱分析,结果色谱图如图1所示。
图1 6号残留溶剂气相色谱图
样品按1.3处理,用顶空进样器取样,上机进行气相色谱质谱联用分析,以质谱全扫描Scan方式得到质谱图,与标准谱库(NIST08)进行相似度检索,结果如表1,确认其中的组分,分别是:2-甲基戊烷、3-甲基戊烷、正己烷、2,2-二甲基戊烷、2,4-二甲基戊烷、甲基环戊烷、2-甲基己烷、环己烷。
图2和图3为2个大豆原油样品残留溶剂的气相色谱图。
表2中为有代表性的大豆原油样品中残留溶剂各组分的百分含量。
表1残留溶剂各组分的相似度检索结果
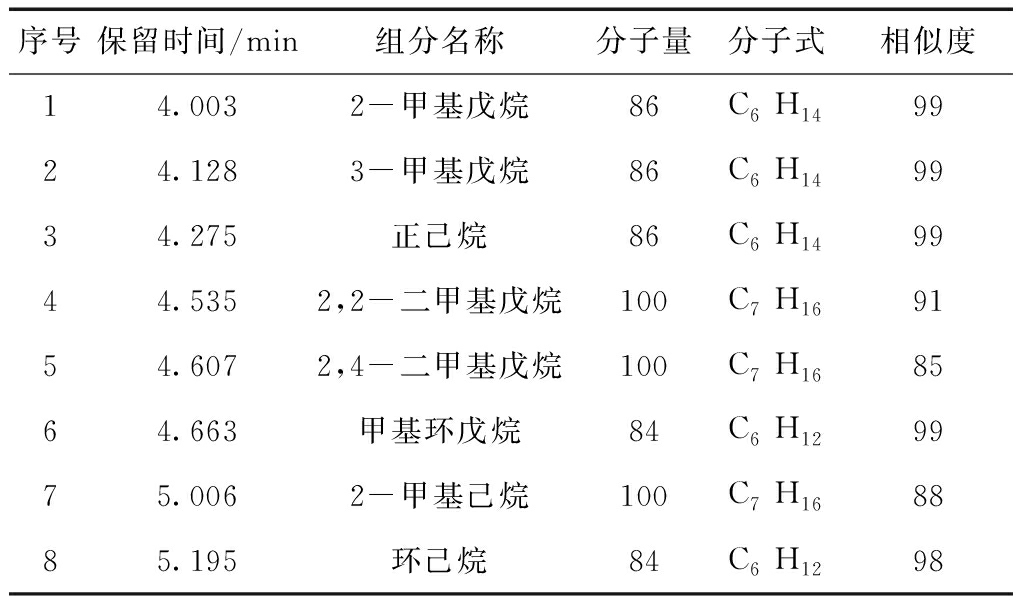
从图2和图3及表2中可以看出:残留溶剂组分2-甲基戊烷、3-甲基戊烷、正己烷、甲基环戊烷以及环己烷在不同大豆原油样品中所占比例差异显著。 2-甲基戊烷所占比例范围在2.76%~35.77%之间;3-甲基戊烷在8.08%~26.80%之间;正己烷在33.29%~47.34%之间;甲基环戊烷在2.45%~38.12%之间;环己烷在0.22%~10.30%之间。各同组分含量值的相对偏差在3.35%~9.78%之间。残留溶剂组分2,2-二甲基戊烷、2,4-二甲基戊烷及2-甲基己烷所占比例变化范围小,各同组分含量值的相对偏差在0.058%~0.12%之间,各组分含量比例均在1%以 下,2,2-二甲基戊烷所占比例范围在0.52%~0.99%之间;2,4-二甲基戊烷所占比例范围在0.22%~0.58%之间; 2-甲基己烷所占比例范围在0.06%~0.23%之间。

图2 样品1气相色谱图

图3 样品2气相色谱图
表2残留溶剂各组成的面积百分含量
%

准确称量2-甲基戊烷、3-甲基戊烷、正己烷、甲基环戊烷、环己烷及正庚烷各100 mg于10 mL容量瓶中,加N-N二甲基乙酰胺定容至刻度。准确吸取各100 μL,分别加入装有5.0 g N-N二甲基乙酰胺及5.0 g新鲜压榨大豆油的20 mL顶空进样瓶中,封盖混合均匀,放入顶空进样器进行气相色谱分析。分别以N-N二甲基乙酰胺和新鲜机榨大豆油作基质,配制各组分的标准混合溶液,测定各组分峰面积A1和峰面积A2,计算各组分的相对质量校正因子f1和f2,结果如表3。
表3各组分相对质量校正因子
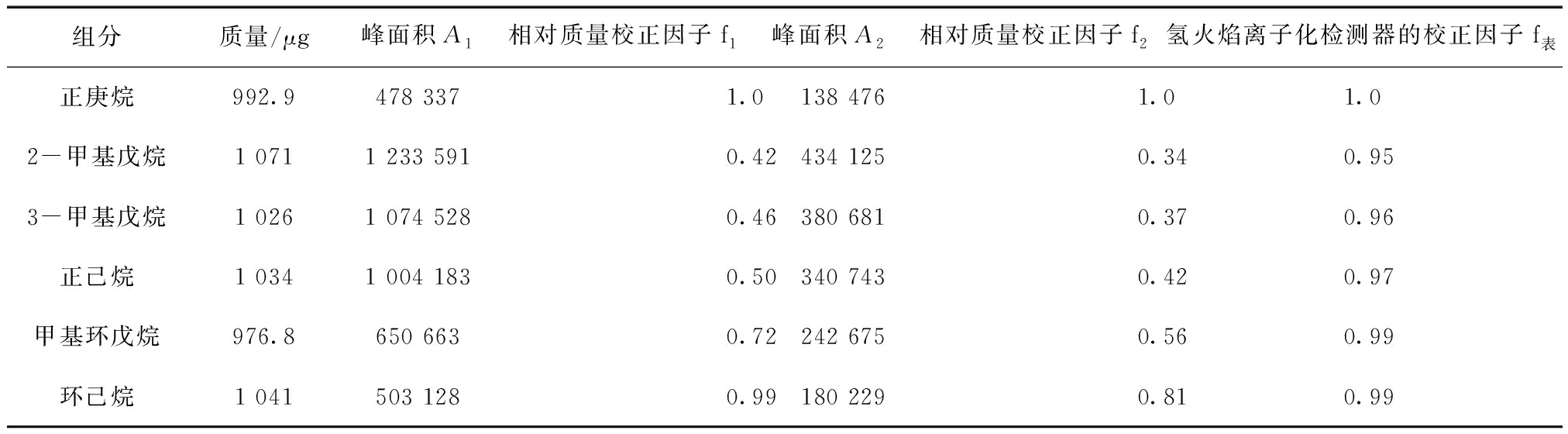
查表[9]氢火焰离子化检测器的校正因子f表,如表3。各组分相对于正庚烷的相对质量校正因子在0.95~0.99之间;以N-N二甲基乙酰胺作基质时各组分的相对质量校正因子在0.42~0.99之间;以新鲜机榨大豆油作基质时各组分的相对质量校正因子在0.34~0.81之间。
以1.4.1配制的6号溶剂为标样,按国标GB 5009.262—2016《食品中溶剂残留量的测定》进行测定[3],以6号溶剂浓度为横坐标,5个组分峰面积总和为纵坐标,得到标准曲线。采用多个峰的峰面积归一法,是基于FID检测六碳烷烃相对质量响应值比较接近,可以认为相对响应因子相同。但本研究发现,残留溶剂内在组成的差异,各组分气相提取效率不同,各组分在顶空气中分配系数存在差异,各组分的相对质量校正因子差异很大,采用峰面积归一法测定的结果可能存在偏差。
用两种方法测定10个不同溶剂残留量的大豆原油样品,测定结果作配对 t检验,以确认两种测定方法结果间是否存在显著性差异。方法1以1.4.2配制的混合溶剂为标样,以每个组分浓度为横坐标,其对应的峰面积为纵坐标,外标法分别定量样品中5个组分含量,样品结果以这5个含量的总和报出,各组分标准曲线方程及相关系数见表4。方法2按国标方法测定,6号溶剂标准曲线方程及相关系数见表4。
表4各组分标准曲线线性方程

两种测定方法比较实验结果数据及计算结果如表5。
表5两种测定方法比较实验结果

计算差值的算术平均值,y-=24.5
计算差值的标准偏差,Sy=14.58
计算t值,![]() 查t表(0.05,,9)=2.26,t计算>t表(0.05,9),两种测定方法结果差异存在统计学意义。可见,应用这两种测定方法会得到不同的测定结果,且结果值越大,测定结果的绝对差值越大。配对t检验结果进一步证实了用峰面积归一法测定食用油中溶剂残留量存在偏差,应谨慎应用。
查t表(0.05,,9)=2.26,t计算>t表(0.05,9),两种测定方法结果差异存在统计学意义。可见,应用这两种测定方法会得到不同的测定结果,且结果值越大,测定结果的绝对差值越大。配对t检验结果进一步证实了用峰面积归一法测定食用油中溶剂残留量存在偏差,应谨慎应用。
各个生产厂家的6号溶剂随来源产地、溶剂循环使用的次数、生产工艺等不同,其溶剂的组成基本相似,主要为2-甲基戊烷、3-甲基戊烷、正己烷、甲基环戊烷、环己烷5个成分,但各组分相对含量差异很大。各组分在顶空气中分配系数不同,各组分的相对质量校正因子差异很大,配对t检验进一步验证了用峰面积归一法测定食用油中溶剂残留量存在偏差。采用各组分的标准溶液以外标法分别定量后加和计算的方法 ,反映了样品中残留溶剂的内在组成及各组分标准溶液的响应差异,可实现对不同批次样品的准确测定。
参考文献:
[1]钱小妹.食用植物油残留溶剂测定方法探讨[J].江苏大学学报(医学版),2003,13(2):166-167.
[2]GB 1535—2003,大豆油[S].
[3]GB 5009.262—2016,食品安全国家标准 食品中溶剂残留量的测定[S].
[4]王姣姣,王宏磊,舒风.HS-GC-MS测定食用植物油中残留溶剂的主要成分[J].广东化工,2016,43(9):220-222.
[5]钱凯,胡建华,赵维佳.食用植物油中溶剂残留的测定方法[J].光谱实验室,2006,23(3):468-471.
[6]王少伟.顶空气相色谱-质谱联用法测定食用植物油中的溶剂残留[J].食品安全导刊,2015,21(3):98-100.
[7]于渤,张素春.不同产地的六号溶剂对溶剂残留测定结果的影响[J].粮食储藏,1997,26(2):49-50.
[8]袁毅,汪海峰.食用油中溶剂残留测定的若干影响因素分析[J].中国油脂,2006, 31(9):59-63.
[9]夏玉宇. 化验员实用手册[M].北京:化学工业出版社,1999, 985-991.
ZHANG Qing-ling
(Fujian Cereals & Oil Quality Supervision and Inspection Institute, Fuzhou Fujian 350002)
Abstract:The composition of 6# residual solvent in soybean crude oil and their area percentages were analyzed and determined by gas chromatograph combined with gas chromatography-mass spectrometry, which indicated that the difference of the ratio between each batches of the soybean crude oil was significant. The fresh extracted soybean oil was used as the basis material to compound standard mixed solution of component to analyze and calculate the relative mass correction factor between 0.34~0.81. The residual solvents in the different batches of the samples can be precisely measured by using different component as standard solution and applying external standard method to quantitative each component then summation calculation, and verified by paired t-test.
Key words:soybean crude oil;residual solvent;composition and ratio;relative mass correction factor;paired t-test
收稿日期:2017-11-10
中图分类号:TS 227
文献标识码:A
文章编号:1007-7561(2018)04-0040-05
作者简介:张青龄,1968年出生,女,高级工程师.