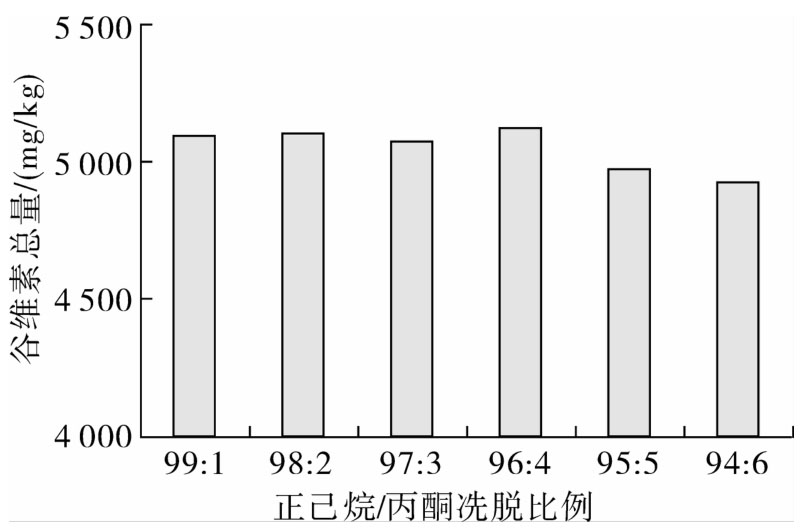
图1 同一样品不同洗脱比例谷维素总量测定值(n=3)
朱 琳1,薛雅琳1,张 东1,柴 杰1,2,安 骏3,刘俊含4
(1.国家粮食局科学研究院,北京 100037;2.江南大学食品学院,江苏无锡 214122;
3.中粮食品营销有限公司,北京 100020;4.北京医院,北京 100730)
摘 要:建立固相萃取柱—液相色谱法测定米糠油中谷维素六种谷维素组分及总量的检测方法。采用二醇基固相萃取小柱提取净化样品,以谷维素混合标准品(γ-Oryzanol)进行定量,色谱柱为Agilent ZORBAX Eclipse Plus C18(250 mm×4.6 mm,5 μm),流动相为含1%乙酸的乙腈∶异丙醇=95∶5,紫外检测器检测(波长321 nm)。六种谷维素组分及总量的平均回收率分别为95.6%~105.7%、97.8%~100.2%,精密度分别为0.1%~4.6%、0.7%~3.4%。该方法可用来测定米糠油中谷维素组分及总量,为深入研究谷维素提供技术支持。
关键词:谷维素;高效液相色谱;米糠油;阿魏酸酯
谷维素是以阿魏酸为骨架的一类酯类混合物,其阿魏酸羧基与环木菠萝醇同系物、植物甾醇生成酯键,从而形成了不同种类的谷维素组分,其中以环木菠萝烯醇阿魏酸酯和24-亚甲基环木菠萝烯醇阿魏酸酯两种谷维素组分为主[1]。谷维素中各阿魏酸酯组分形成了其诸多的生理功能,如抗氧化、调节植物神经功能、降低血清胆固醇、治疗高血压等生理活性[2]。米糠油中富含谷维素,是人体摄入天然谷维素的重要来源,但因谷维素中各种阿魏酸组分对热的稳定性不同,在毛米糠油精炼过程中,谷维素组分会发生结构的变化,其受热后酯键、双键和甲氧基是分解的主要对象,首先从阿魏酸酯部分开始,再至环木菠萝醇和甾醇部分[3]。因此,有必要建立一种测定米糠油加工精炼前后谷维素组分及总量变化的检测方法。
目前,研究报道植物油中谷维素分析方法较多,针对植物油谷维素总量的测定方法有紫外分光光度法、高效液相色谱氨基柱法等[4-5],但这些方法仅能测谷维素总量,不能有效地分离定量谷维素组分;针对谷维素组分的测定方法主要是高效液相色谱C18柱法,通常采用甲醇∶乙腈∶异丙醇=45∶50∶5[6-7]作为流动相,在此条件下会随着分析次数的增加,谷维素组分的分离度会逐步下降;针对植物油中谷维素组分及总量测定的前处理主要是去除植物油中的甘三酯成分,相关报道有用乙腈∶甲醇∶异丙醇=50∶45∶5[8]或异丙醇∶乙腈=70∶30)[9]作为提取溶剂,但这两种方法回收率较低,且由于油中甘三酯会溶于异丙醇,使得净化不完全,也有文献报道用硅胶固相萃取柱进行净化处理[6],笔者将净化处理后样品进行甘三酯检测,仍会有甘三酯残留,净化并不完全。本研究利用二醇基(Diol)固相萃取柱对米糠油进行净化处理后,不仅回收率高,且净化完全,采用液相色谱—质谱联用仪Q-TOF定性分析出米糠油中六种主要谷维素组分,再利用高效液相色谱法对这六种谷维素组分及总量进行定量分析,该方法不会随着分析次数的增加而使各谷维素组分分离度下降,为今后研究米糠油精炼前后谷维素组分及总量的变化提供了检测依据,并为科学评价米糠油的营养价值提供技术支撑。
1.1 材料与试剂
二醇基(Diol)固相萃取柱(500 mg/6 mL):博纳艾杰尔科技有限公司;乙酸、异丙醇、乙腈、正己烷、丙酮均为色谱纯:赛默飞世尔科技公司;环木菠萝烯醇阿魏酸酯(纯度≧99%):日本和光纯药工业株式会社(WAKO);谷维素混合标准品(γ-Oryzanol):东京化成工业株式会社;化学精炼、物理精炼毛米糠油和成品米糠油:工厂精炼车间取样。
1.2 主要仪器设备
1100 型高效液相色谱仪(带UV检测器):美国安捷伦(Agilent)公司;1200型高效液相色谱仪串联6510型四极杆飞行时间质谱仪:美国安捷伦(Agilent)公司;固相萃取装置:博纳艾杰尔科技有限公司。
1.3 样品前处理
二醇基固相萃取柱:加20 mL正己烷活化。
测试溶液:称取0.5 g油样于10 mL刻度试管瓶中,用正己烷定容至5 mL。
净化:取50~300 μL测试溶液(上柱的谷维素总量不超过150 μg)于已经活化的二醇基固相萃取柱中,加入5 mL正己烷进行淋洗,弃去全部的流出液,再加入3 mL正己烷/丙酮(96/4)混合溶液淋洗萃取柱,待混合淋洗液全部通过后立即抽干萃取柱,弃去全部的流出液。分三次加入4、2、2 mL丙酮洗脱固相萃取柱,每次洗脱后均需抽干固相萃取柱,三次洗脱液收集在10 mL试管中,用氮气吹干丙酮后,用0.5~1 mL丙酮将该试管壁的残渣洗于试管底部,再次用氮气吹干;加入1 mL异丙醇超声溶解1 min涡旋混匀后,用0.45 μm滤膜将样品溶液过滤至棕色进样瓶中,待测。
1.4 谷维素组分质谱定性分析条件
色谱部分。色谱柱:Agilent ZORBAX Eclipse Plus C18(250 mm×4.6 mm,5 μm);柱温:40℃;流速:0.6 mL/min;进样量:20 μL;流动相为乙腈∶异丙醇=90∶10,流动相中加入终浓度为10 mmoL/mL醋酸铵。
质谱部分。采集模式:大气压化学电离源(APCI)负离子模式;干燥气流速:350℃;干燥气流速:4 L/min;气化器温度:350℃;毛细管电压:3.0 kV;电晕电流:15 μA;全扫描范围:m/z 500~700;子离子扫描范围:m/z 100~700。
1.5 谷维素组分液相定量色谱条件
色谱柱:Agilent ZORBAX Eclipse Plus C18(250 mm×4.6 mm,5 μm);柱温:30℃;流速:1.0 mL/ min;进样量:20 μL;紫外检测器波长:321 nm;流动相:含1%乙酸的乙腈∶异丙醇=95∶5。
2.1 净化方法选择
植物油样品的净化主要是去除样品中的甘油三酯成分,采用二醇基固相萃取柱通过不同淋洗液比例对样品进行净化处理,净化处理后谷维素总量见图1。从图1可以看出,99∶1、98∶2、97∶3、96∶4淋洗比例对谷维素总量的测定影响并不大,相对标准偏差为0.42%。这四个淋洗比例净化后样品中甘三酯残留检测采用AOCS甘三酯示差方法测定[10],净化前后待测溶液中甘三酯残留情况见图2。从图2可以看出,淋洗液比例在正己烷∶丙酮=96∶4时,其内几乎无甘油三酯成分残留。因此,正己烷∶丙酮= 96∶4为最佳淋洗液比例。
图1 同一样品不同洗脱比例谷维素总量测定值(n=3)

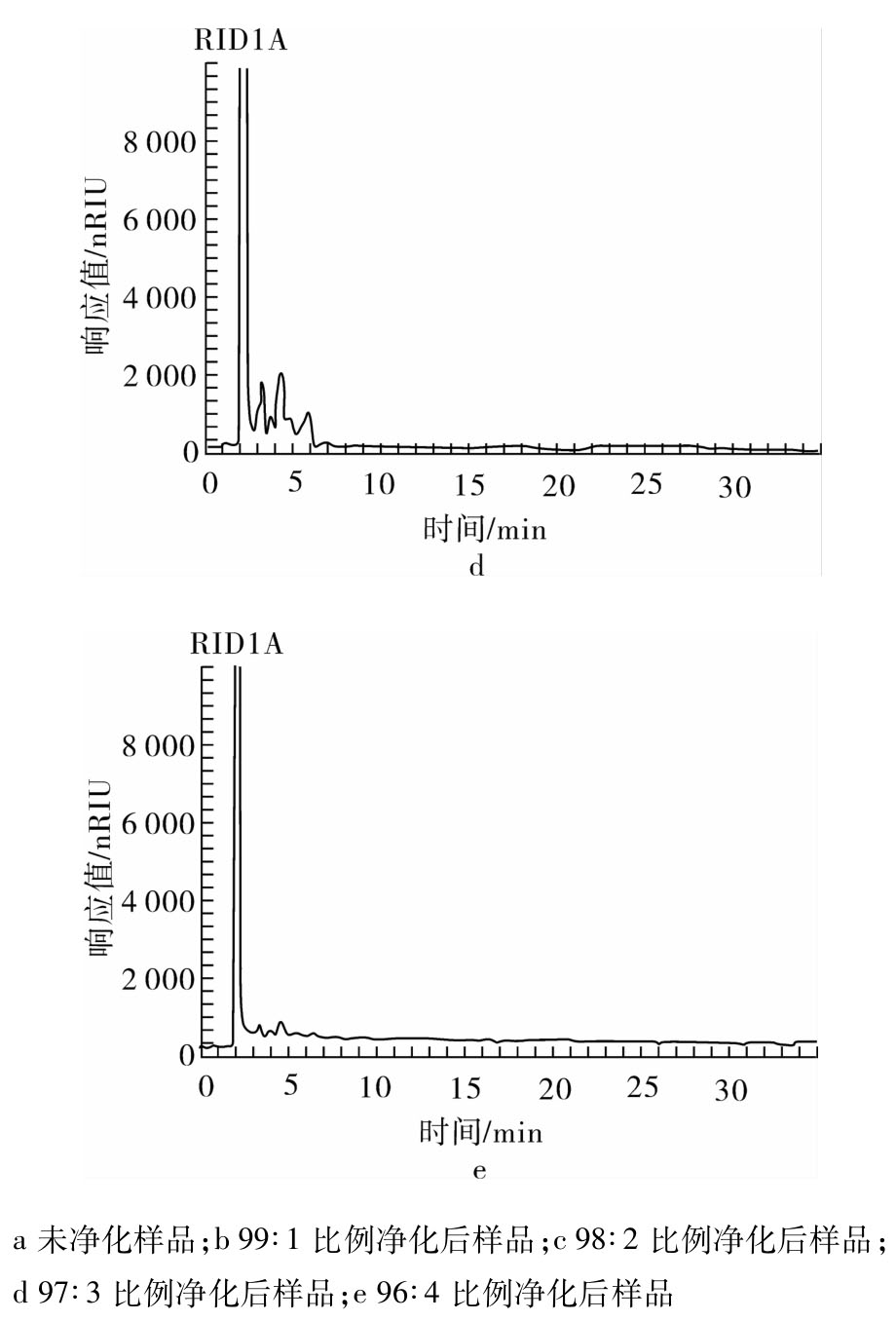
图2 净化前与四种比例淋洗液净化后样品的甘油三酯残留
2.2 质谱定性分析
采用液相色谱—质谱联用仪Q-TOF定性分析米糠油中谷维素组分,经C18色谱柱洗脱分离。六种谷维素组分分子式、保留时间、精确质量数、质量偏差及碎片离子见表1。从表1可以看出,各组分的理论分子量与实际测定分子量的相对偏差均小于10 ppm,同时,通过二级质谱得到的碎片离子可以看出,586.397 2、600.417 9、560.385 2、574.403 0、588.417 6以及576.417 4为谷维素失去CH3得到的碎片离子,而133、193、175分别为阿魏酸失去COOH、H、H2O得到的碎片离子。分析结果与Rogers E J等文献[7-8,11]基本一致,从而确定为谷维素的六种组分。
表1 六种谷维素组分的相关参数

2.3 高效液相色谱定量分析
2.3.1 色谱条件的选择
采用常用的甲醇∶乙腈∶异丙醇=45∶50∶5作为流动相分析谷维素组分时,谷维素组分会随着分析次数的增加分离度逐步降低。通过改进流动相即在乙腈中加入1%乙酸会使谷维素组分洗脱下来,不会导致分离度的下降,再加5%异丙醇可加快洗脱速度。谷维素标准品及米糠油样品中谷维素组分液相色谱图见图3。
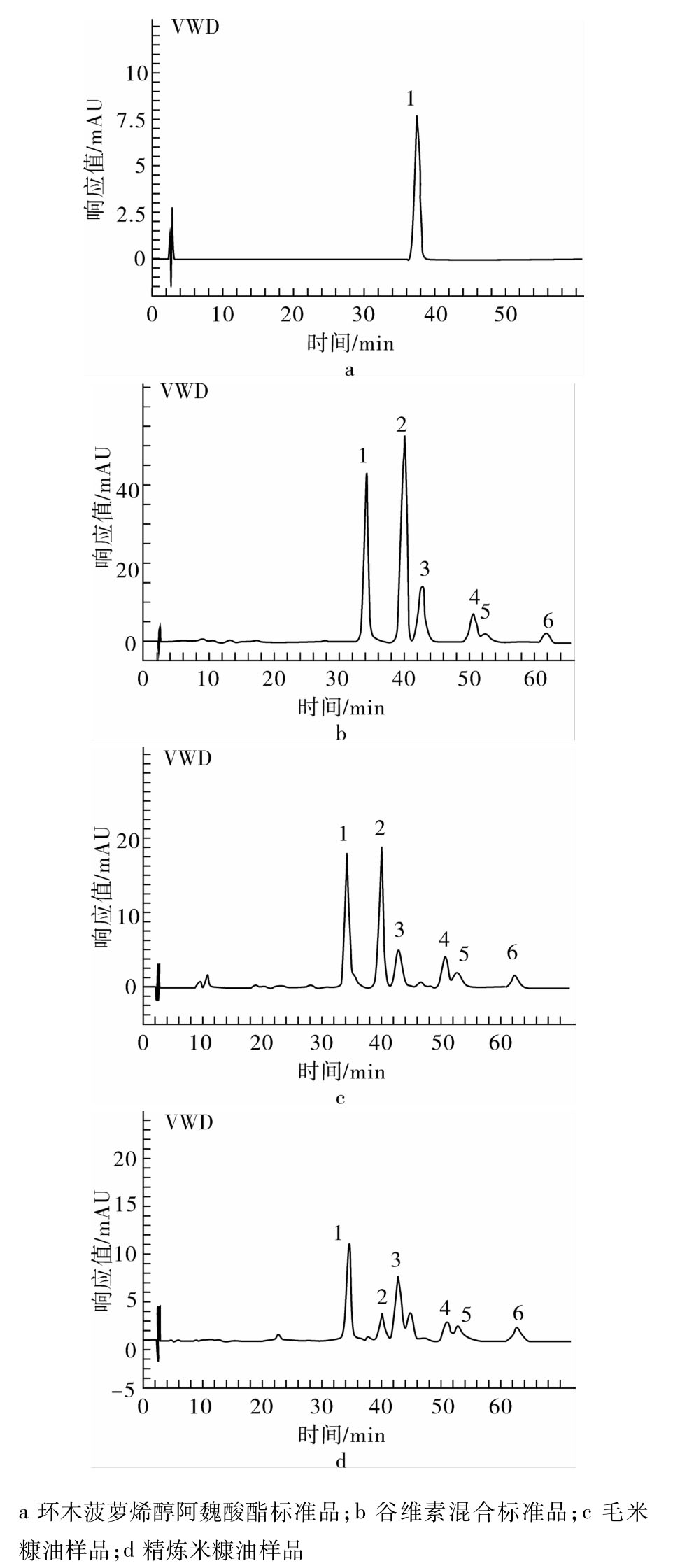
图3 谷维素标准品及米糠油样品中谷维素组分液相色谱图
注:峰1,环木菠萝烯醇阿魏酸酯;峰2,24-亚甲基环木菠萝烯醇阿魏酸酯;峰3,菜油甾醇阿魏酸酯;峰4,β-谷甾醇阿魏酸酯;峰5,环木菠萝醇阿魏酸酯;峰6,二氢谷烷醇阿魏酸酯。
2.3.2 标准品的选择、线性范围和检出限
通过面积归一化法,采用谷维素混合标准品对各谷维素组分进行定量。把所有的出峰面积(除溶剂峰外)看作100%,峰1至峰6的峰面积占(除溶剂峰外)总峰面积的百分数即为谷维素混合标准品的纯度。本研究此次选用的谷维素混合标准品利用面积归一化法计算得其纯度为96.4%,组分1至组分6在混合标准品的百分含量分别为:31.45%,43.60%,15.33%,7.20%,2.10%,0.32%。
以异丙醇为溶剂,配制1 000 μg/mL的谷维素混合标准储备液,进一步稀释成5、10、20、50、100、200 μg/mL的谷维素混合标准工作溶液,以浓度(x)为横坐标、峰面积(y)为纵坐标作图得到线性方程,再以S/N≧3确定方法的检出限,谷维素六种组分及总量的线性方程及检出限见表2。
配制200 μg/mL组分1(环木菠萝烯醇阿魏酸酯)的单标准储备液,进一步稀释成1、10、20、50、100 μg/mL工作溶液,用异丙醇超声溶解定容,以浓度(x)为横坐标、峰面积(y)为纵坐标作图得到线性方程,再以S/N≧3确定方法的检出限,谷维素组分1(环木菠萝烯醇阿魏酸酯)的线性方程及检出限见表3。
从表2~表3不仅可以看出各组分和总量的线性方程和检出限,同时可以看出,利用混合标准品面积归一化法计算组分1的线性方程和组分1单标的线性方程的斜率基本一致,相关性因子为0.99。因此,利用混合标准品和单标对组分1进行定量基本是一致的。据文献[6]报道,谷维素各组分为同系物,其相关因子为1,因此,可以采用混合标准品对样品中的谷维素进行定量分析。
表2 谷维素混合标准品六种组分及总量的线性方程和检出限
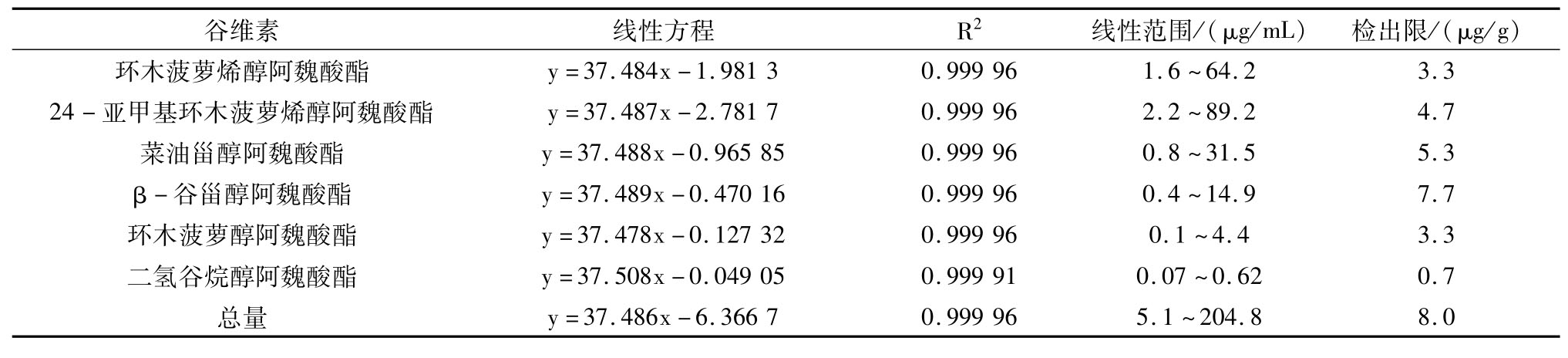
表3 谷维素单标环木菠萝烯醇阿魏酸酯的线性方程和检出限

2.3.3 方法的加标回收率和精密度
在不含谷维素的大豆油中分别添加不同浓度(484.1、5 375.8、9 929.2 mg/kg)的谷维素混合标准品,进行回收率和精密度实验。结果如表4所示,不同水平谷维素各组分的平均回收率为95.6%~105.7%,相对标准偏差为0.1%~4.6%,能满足六种谷维素组分的测定要求,不同水平谷维素总量的平均回收率为97.8%~100.2%,相对标准偏差为0.7%~3.4%。
表4 回收率和精密度检测结果(n=2)
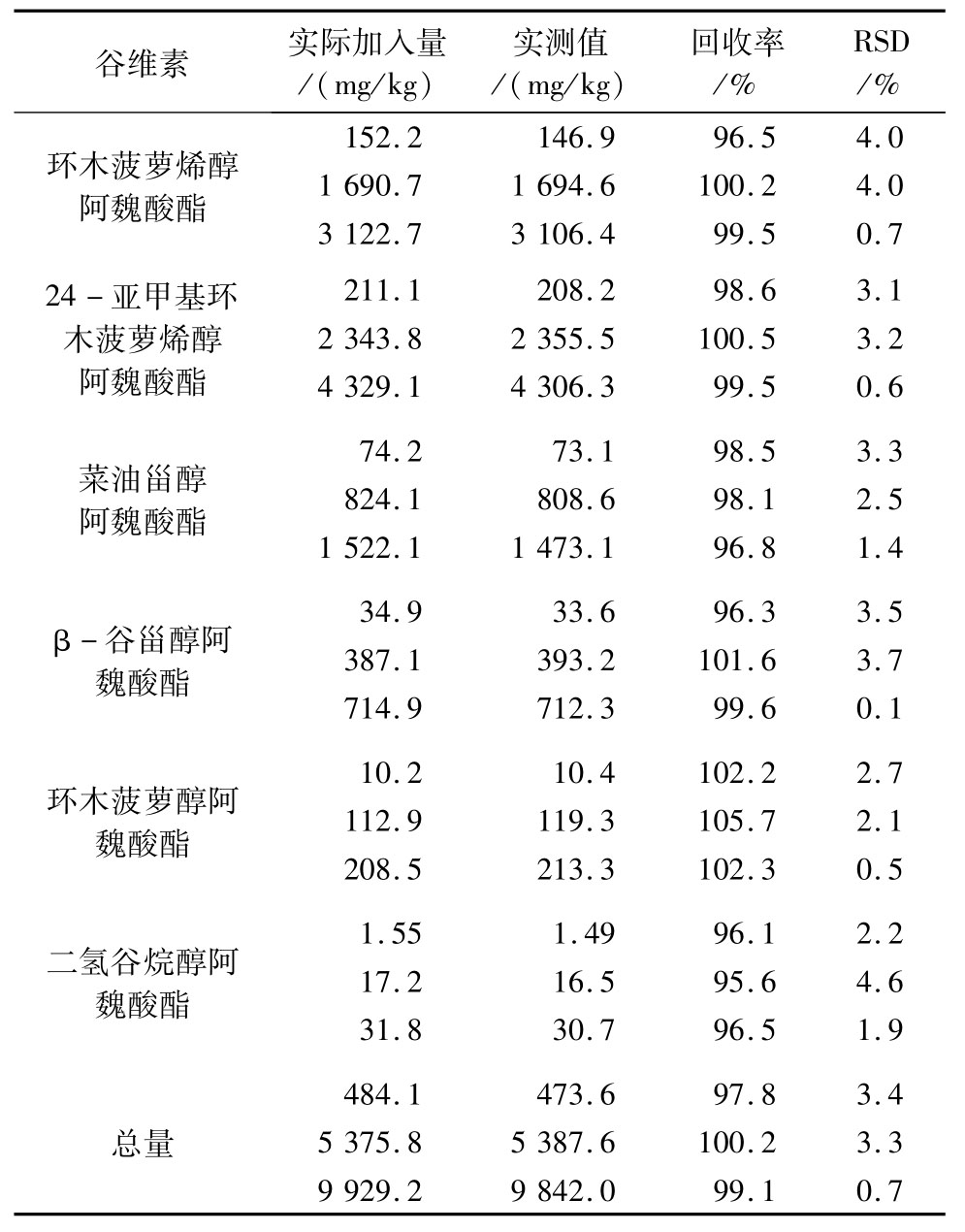
2.3.4 实际样品的测定
分别取化学精炼前后的米糠油和物理精炼前后的米糠油共计四个样品,利用本方法对其谷维素组分及总量进行测定分析,结果见图4。
从图4可以看出,化学精炼米糠油谷维素总量保留了8.54%,组分3保留最高(20.76%),谷维素组分2仅保留了2.75%。物理精炼较化学精炼米糠油谷维素保留量高,除组分2较精炼前略有升高外,谷维素总量和其他组分均保留了70%左右。从整体上看,物理精炼的米糠成品油要比化学精炼的米糠成品油谷维素保留量高出7倍。这可能是由于加工目的不一致导致的,化学精炼的米糠油可得到生产谷维素药品的副产物,物理精炼的米糠油可将谷维素尽可能多的保留在油脂中。但不管化学精炼还是物理精炼,谷维素组分及总量均会有不同程度的损失,通过本方法可知谷维素组分及总量具体保留量,为今后加工前后谷维素组分的深入研究提供检测依据。
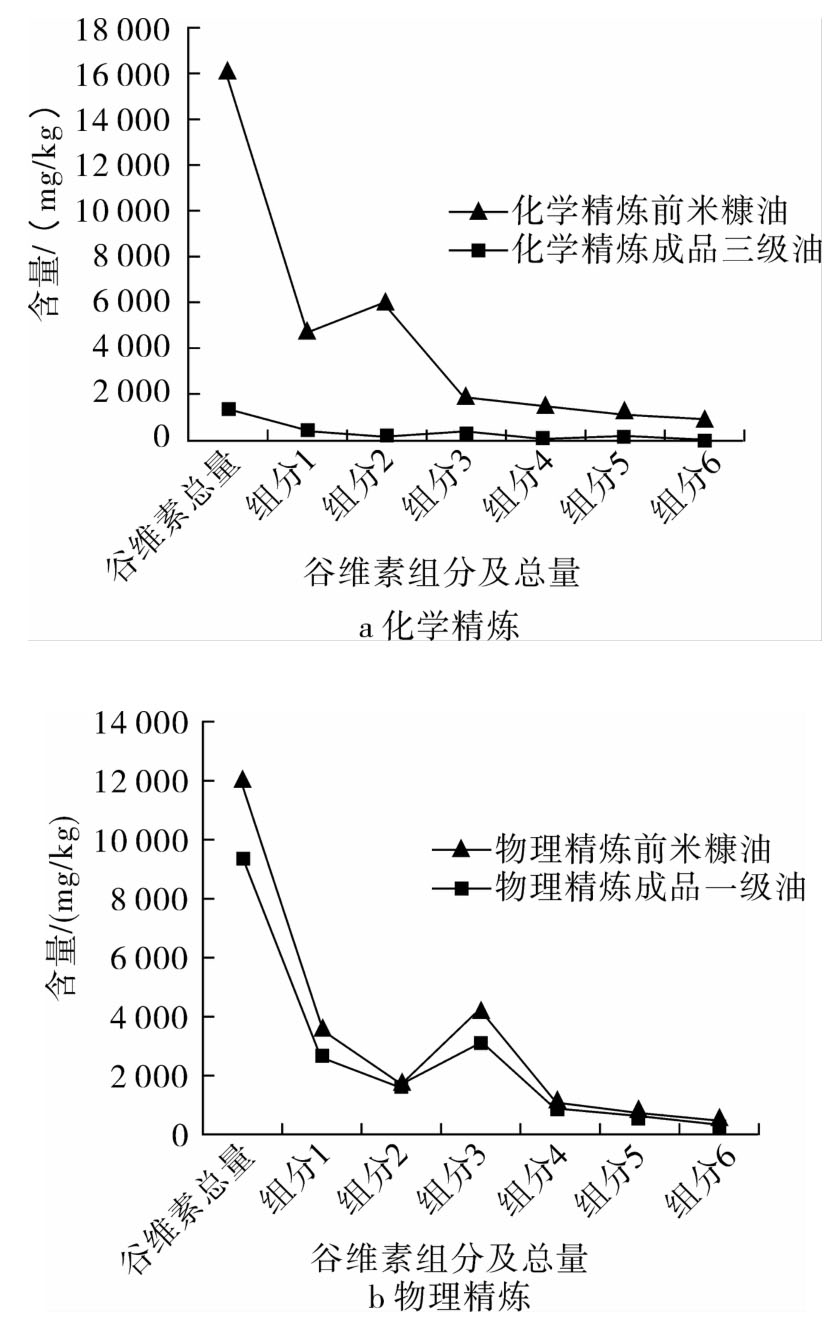
图4 精炼前后谷维素组分及谷维素总量变化情况
本研究建立了二醇基固相萃取柱对米糠油进行净化处理,混合标准品定量谷维素组分及总量的高效液相色谱法。该方法不仅可将米糠油中污染色谱柱的甘油三酯成分基本去除,同时,谷维素组分及总量的平均回收率可分别达到95.6%~105.7%和97.8%~100.2%,精密度可分别达到0.1%~4.6%和0.7%~3.4%。由此可以得出,该方法样品前处理省时简便、成本低廉、回收率高。该方法的建立可为谷维素组分的深入研究提供检测依据,同时,为科学评价米糠油营养成分提供技术支撑。
参考文献:
[1]吴素萍.谷维素的生理功能及提取方法的研究现状[J].食品工业科技,2009,30(8):365-368.
[2]田媛媛,林亲录,罗非君.米糠γ-谷维素生理功能研究新进展[J].粮食与油脂,2013,26(6):43-46.
[3]刘大川,李从军.米糠油的营养特性及精炼新工艺[J].中国油脂,2014,39(2):13-16.
[4]范国欣,孙晓薇,潘希荣.米糠油中谷维素含量的测定[J].粮油食品科技,2010,18(1):20-21.
[5]陈燕飞,金俊,郑苏卫,等.高效液相色谱法测定米糠油中谷维素含量[J].中国粮油学报,2014(7):112-115.
[6]LU W Y,NIU Y G,YANG H S,et al.Simultaneous HPLC quantification of five major triterpene alcohol and sterol ferulates in rice bran oil using a single reference standard[J].Food Chemistry,2014,148:329-334.
[7]龚院生,姚惠源.γ-谷维醇的液质联用分析[J].化学世界,2001,42(3),123-125.
[8]ROGERS E J,RICE S M,NICOLOSI R J,et al.Identification and quantitation of γ-oryzanol components and simultaneous assessment of tocols in rice bran oil[J].Journal of American Oil Chemists`Society,1993,70(3):301-307.
[9]PESTANA V R,ZAMBIAZI R C,MENDONCA C R B,et al.Quality Changes and Tocopherols and γ-Orizanol Concentrations in Rice Bran Oil During the Refining Process[J].J Am Oil Chem Soc,2008,85:1013-1019.
[10]AOCS Official Method Ce 5b-89.Triglycerides in vegetable oils by HPLC[S].
[11]KIM H W,KIM J B,SHANMUGAVELAN P,et al.Evaluation of γ-oryzanol content and composition from the grains of pigmented rice-germplasms by LC-DAD-ESI/MS[R].Bmc Research Notes,2013,6:149-159.
Determination of oryzanol in rice bran oil by high performance liquid chromatography
ZHU Lin1,XUE Ya-lin1,ZHANG Dong1,CAI Jie1,2,AN Jun3,LIU Jun-han4
(1.Academy of State Administration of Grain,Beijing 100037;
2.School of Food Science and Technology,Jiangnan University,Wuxi Jiangsu 214122;
3.COFCO Food Sales&Distribution Co.,Ltd,Beijing 100020;
4.Beijing Hospital,Beijing 100730)
Abstract:An analytical method based on solid phase extraction-high performance liquid chromatography was established for the determination of six oryzanol compositions and total oryzanol content in rice bran oil.The samples were purified by diol-silica gel series SPE column.The standard sample(γ-Oryzanol)was used for quantification.The chromatographic column was Agilent ZORBAX Eclipse Plus C18(250 mm×4.6 mm,5 μm)and the mobile phase was acetonitrile with 1%acetic acid∶isopropanol= 95∶5.The wavelength of UV was 321 nm.The average recoveries of six compositions and total content were in the range of 95.6%~105.7%and 97.8%~100.2%with relative standard deviations of 0.1%~4.6%and 0.7%~3.4%.The method was suitable for determination of oryzanol composition and content in rice bran oil,which provided technical support for further research of oryzanol.
Key words:oryzanol;high performance liquid chromatography;rice bran oil; ferulic acid ester
中图分类号:TS 225.1
文献标识码:A
文章编号:1007-7561(2016)05-0038-06
收稿日期:2015-12-08
基金项目:碾米制粉制油节粮节能智能新装备研发与示范(201513003)
作者简介:朱琳,1984年出生,女,助理研究员.
通讯作者:薛雅琳,1960年出生,女,教授级高级工程师.