刘世娟,张海弢, 王 婧, 徐振秋
(江苏康缘药业股份有限公司, 江苏 连云港 222001)
摘 要:建立一种辅助降血糖保健食品中水溶性多糖的测定方法,考察提取方式、醇沉浓度、脱蛋白方法及显色方法等因素对多糖测定结果的影响。结果表明选用苯酚硫酸法进行显色,超声提取60 min,超声功率250 W,醇沉浓度80%,以样液体积1/10倍量的5%三氯乙酸溶液,脱蛋白1次,静置时间2 h,测定结果较好。葡萄糖浓度在6.61~79.28 μg/mL范围内与吸光度有良好的线性关系,回归方程为A=0.014 4C-0.065 5(r=0.999 6),平均加标回收率为98.47%,RSD为1.64%。测得样品中水溶性多糖平均含量为11.49%,RSD为2.31%。 该方法简便、准确,重复性好,可用于辅助降血糖保健食品中多糖含量的测定和质量控制。
关键词:水溶性多糖;测定方法;苯酚硫酸法;保健食品
多糖是指十个以上单糖分子通过糖苷键连接而成的碳水化合物,大多具有调节免疫、抗肿瘤、抗炎、降血糖、降血脂、抗衰老等多方面的生物活性和保健功能,广泛存在于植物(如桑叶、枸杞等)、藻类(如紫菜、海带等)和菌类(如茯苓、虫草等)中。近年来作为保健食品功效成分的研究日益受到重视,其测定方法对于多糖产品的研究和开发有着较为重要的意义,但目前尚无国家标准,常用的有比色法(如苯酚硫酸法、蒽酮硫酸法等)、滴定法(如直接滴定法、间接碘量法等)、色谱法(包括气相色谱法、高效液相色谱法、薄层色谱法)等。滴定法常因水溶性淀粉、糊精等物质的干扰而使灵敏度不高,回收率偏低[1]或在滴定过程中由于碘不可避免的挥发而导致滴定误差[2];色谱法主要用于组成多糖的单糖定量或定性分析,对色谱柱、检测器等要求较高[3]。比色法是一种传统的测定多糖含量的方法,其操作简便、易行且结果准确[4]。本研究参照文献报道及保健食品多糖功效成分检测方法[5-6],结合产品的生产工艺,建立一种辅助降血糖保健食品中水溶性多糖测定方法,结果表明该方法简便,重复性好、准确度高,可作为多糖类保健食品质量控制的依据。
1.1 材料与设备
清平片,以黄芪、玉竹等药用植物为主要原料制成的辅助降血糖保健食品,由江苏康缘药业股份有限公司提供。D-无水葡萄糖,中国食品药品检定研究院,批号为110833-201205,含量以99.5%计;牛血清白蛋白,中国食品药品检定研究院,批号为40619-201120;考马斯亮蓝G-250,国药集团化学试剂有限公司;水为去离子水;三氯乙酸、氯仿、正丁醇、无水乙醇、苯酚、蒽酮、浓硫酸等均为国产分析纯。
UV2700-PC紫外可见分光光度计;KQ-250DB型数控超声波清洗器;TDL-5-A低速大容量离心机;Sartorius BP-211D电子天平;DHG-9145A电热恒温鼓风干燥箱;HWS26电热恒温水浴锅。
1.2 测定方法
多糖含量的测定:苯酚硫酸法[7]、蒽酮硫酸法[8]。
蛋白质含量的测定:考马斯亮蓝法[9]。
1.3 实验方法
1.3.1 多糖含量的测定
1.3.1.1 苯酚硫酸法
以葡萄糖为标准样品,得到标准曲线方程A=0.014 4C-0.065 5(r=0.999 6,n=7),线性范围为6.61~79.28 μg/mL。
样品中多糖含量的测定:精密量取待测液0.2 mL, 置于10 mL具塞刻度比色管中,以去离子水补加至2.0 mL,加入6.25%苯酚溶液1.0 mL,摇匀,冰浴中滴加5.0 mL浓硫酸,摇匀后置沸水浴中加热15 min,冰水中冷却至室温,于490 nm波长处测吸光度值。另以2.0 mL去离子水按上述操作为空白,根据标准曲线得到样品多糖含量。
1.3.1.2 蒽酮硫酸法
以葡萄糖为标准样品,得到标准曲线方程A=0.008 7C -0.080 3(R2=0.999 8,n=6),线性范围为13.21~132.13 μg/mL。
样品中多糖含量的测定:精密量取待测溶液0.1 mL, 置于10 mL具塞刻度比色管中,以去离子水补加至1.0 mL,冰水浴中迅速滴加0.2%蒽酮硫酸溶液5.0 mL,摇匀后置沸水浴中加热10 min,流水中冷却20 min,于620 nm波长下测吸光度值。另以1.0 mL去离子水按上述操作为空白,根据标准曲线得到样品多糖含量。
1.3.2 蛋白质含量的测定
以牛血清白蛋白为标准样品,得到标准曲线方程为:Y=0.007 5X+0.005 5(R2=0.998 8,n=7),线性范围为4.14~82.80 μg/mL。
样品中蛋白质含量的测定:精密量取待测溶液1.0 mL,加入考马斯亮蓝G-250试剂5.0 mL,摇匀,5 min后于595 nm波长处测吸光度,以1.0 mL去离子水按上述操作为空白,根据标准曲线得到样品蛋白质含量。
1.3.3 供试品的制备
样品经除去包衣、研细,过40目筛,取约1 g,精密称定,至三角瓶中,加水50 mL,按不同的处理方法进行提取、放冷、摇匀、离心(3 500 r/min,15 min),上清液移入烧杯中,按不同醇沉浓度加入乙醇,搅拌均匀,置4 ℃冰箱醇沉过夜。离心,弃上清,沉淀物用少量乙醇洗涤、离心,重复2次,用水溶解,转移至100 mL容量瓶中,定容至刻度,摇匀。按不同方法进行脱蛋白处理,取上清液离心,即得供试品溶液。
1.3.4 样品中多糖含量(以无水葡萄糖计)
多糖含量![]() ×稀释倍数×100%。
×稀释倍数×100%。
2.1 单因素对多糖含量测定的影响
2.1.1 提取方式
采用超声(250 W, 40 kHz)提取,比较30 min、60 min的提取效果;采用100 ℃水浴回流提取,比较2、3 h的提取效果。采用沸水浴提取,比较2、3 h的提取效果,结果见图1。
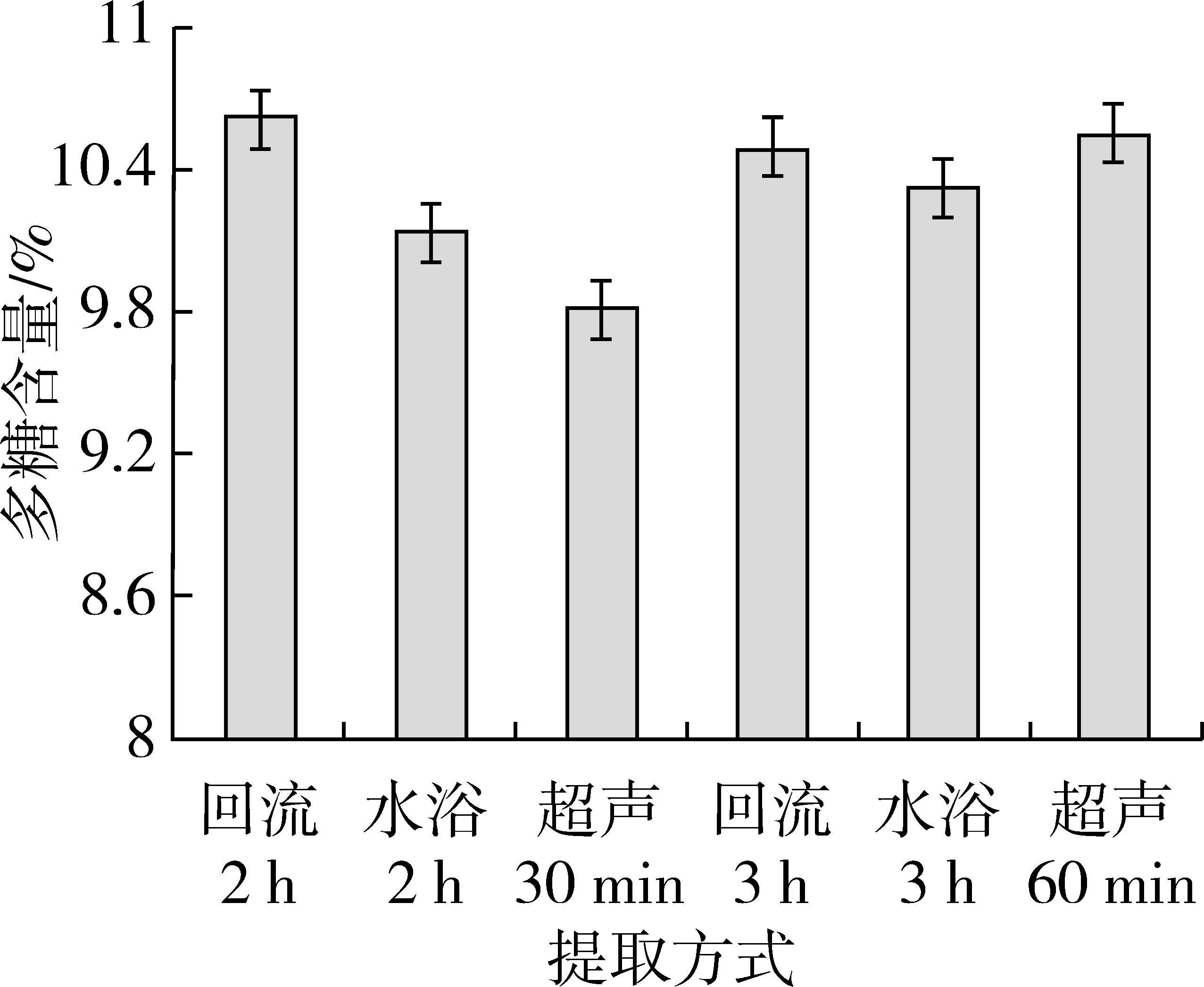
图1 提取方式对多糖含量测定的影响
注:样品醇沉浓度为85%;加入样液体积1/10倍量、5% 三氯乙酸溶液,静置2 h进行脱蛋白处理;苯酚硫酸显色法。
由图1可知,沸水浴与回流提取的多糖含量与时间关系并不明显;超声提取随时间的延长,提取液中多糖含量增加,且超声提取60 min与回流提取2 h的含量结果无明显差异。由于超声提取操作简单、时间短、提取较彻底,故选超声提取60 min为提取方法。
2.1.2 醇沉浓度
加入不同量乙醇,使醇沉终浓度分别达60%、70%、80%、90%和95%,比较不同醇沉浓度对多糖含量的影响,结果见图2。

图2 醇沉浓度对多糖含量测定的影响
注:样品经超声提取60 min;加入样液体积1/10倍量、5% 三氯乙酸溶液,静置2 h进行脱蛋白处理;苯酚硫酸显色法。
由图2可知,醇沉终浓度为80%时的多糖含量最高。因为低浓度乙醇沉淀下来的主要是高分子量的多糖和杂蛋白;中浓度乙醇沉淀下来的是中等分子量的多糖和少量杂蛋白;而高浓度乙醇沉淀下来的是较小分子量的多糖和低聚糖及一些多肽;90%乙醇沉淀的粗多糖含量降低是由于有些多糖分子与醇溶蛋白相结合,使多糖也随之溶于乙醇溶液中,没有沉淀而导致多糖含量降低[10]。故醇沉终浓度选用80%为佳。
2.1.3 脱蛋白方法对比
用水提醇沉法提取多糖时,一般乙醇沉淀的方法会去除部分小分子杂质,而植物中的蛋白质却较多地沉淀出来影响多糖的测定。故测定多糖时应先用蛋白沉淀剂沉淀蛋白,以减少蛋白质对多糖含量测定的影响。
亚铁氰化钾—乙酸锌法:精密吸取供试液6份(各2 mL)于10 mL容量瓶中,分别加入5%、10%、15%、20%、25%、30%体积的21.9%醋酸锌和等量的10.6%亚铁氰化钾,水定容至刻度,摇匀,静置1 h,取上清液离心,分析其多糖和蛋白质含量。
三氯乙酸(TCA)法:精密吸取供试液6份(各2 mL)于10 mL容量瓶中,分别加入浓度为0.5%、2%、3.5%、5%、6.5%、8%的三氯乙酸0.2 mL,水定容至刻度,摇匀,静置2 h,取上清液离心,测其多糖和蛋白质含量。
Sevag法:精密吸取供试液6份(各5 mL)于具塞离心管中,加入1 mL Sevag试剂(氯仿∶正丁醇=4∶1),充分振摇10 min,经3 500 r/min离心15 min,弃下层有机相和中间的变性蛋白质,分取水层,定容至10 mL。6份样品分别处理1、2、3、4、5和6次,分析其多糖和蛋白质含量。
比较三种方法的脱蛋白效果,结果见图3。由图可知,采用亚铁氰化钾—乙酸锌法除蛋白,当试剂的添加量达到样品液的20%时,蛋白质脱除率达78.66%,多糖损失率为34.92%,随着试剂用量的继续增加,蛋白脱除率逐渐下降,而多糖损失率仍继续增加;TCA法除蛋白,增加TCA浓度可以提高蛋白质脱除率,但多糖损失率也在增加,5%的TCA蛋白脱除率为80.22%,多糖损失率18.27%,当TCA浓度继续增加,蛋白质脱除率增加缓慢,而多糖损失率增加幅度较大;Sevag法除蛋白,随着脱蛋白次数的增加,蛋白质脱除率逐渐增加,经过4次脱蛋白处理之后增加趋势稍减弱,经过6次脱蛋白处理后,蛋白质脱除率仅为36.02%,多糖损失率为23.31%。故在兼顾多糖损失率和蛋白质脱除率的情况下,选用5%的三氯乙酸去除蛋白质,并进一步优化。

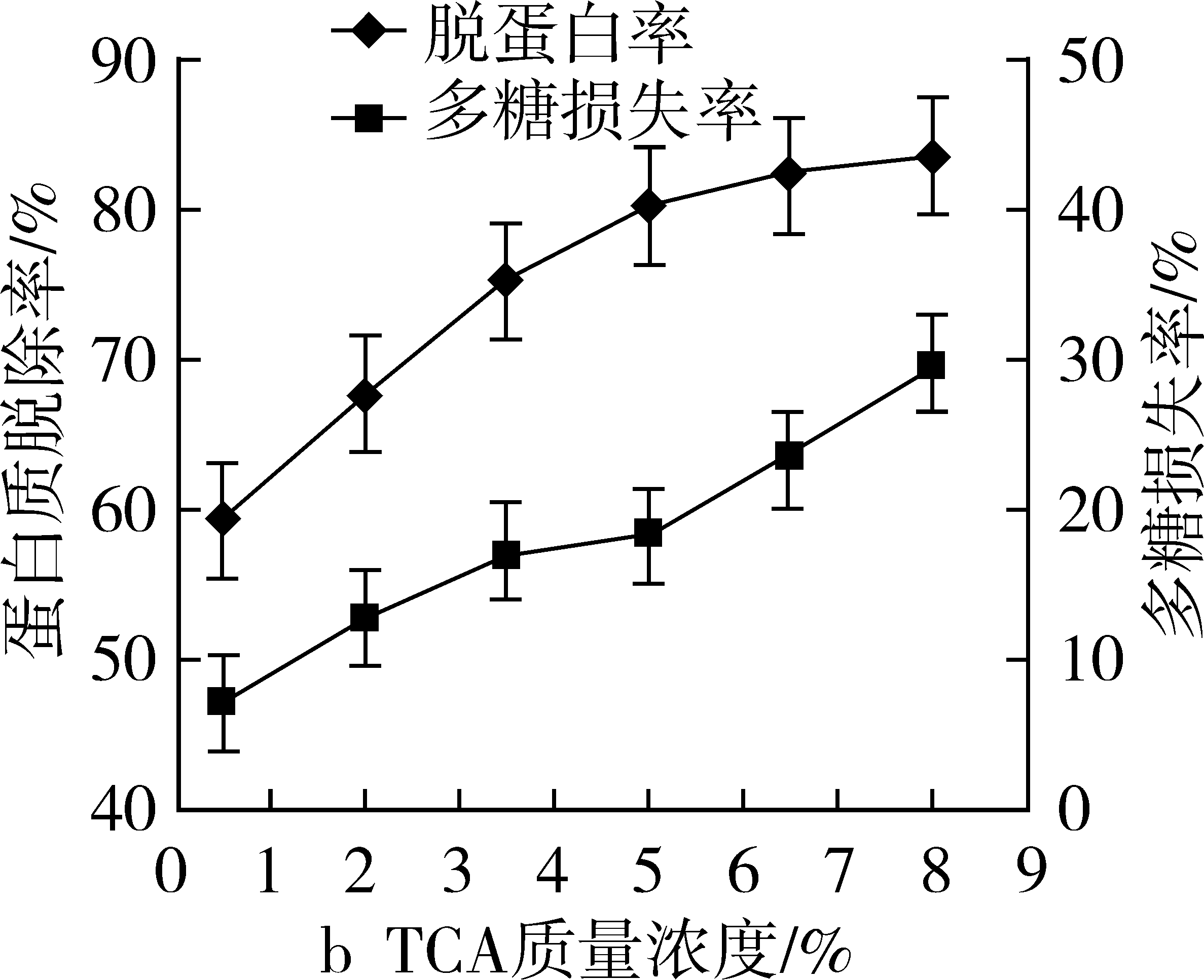
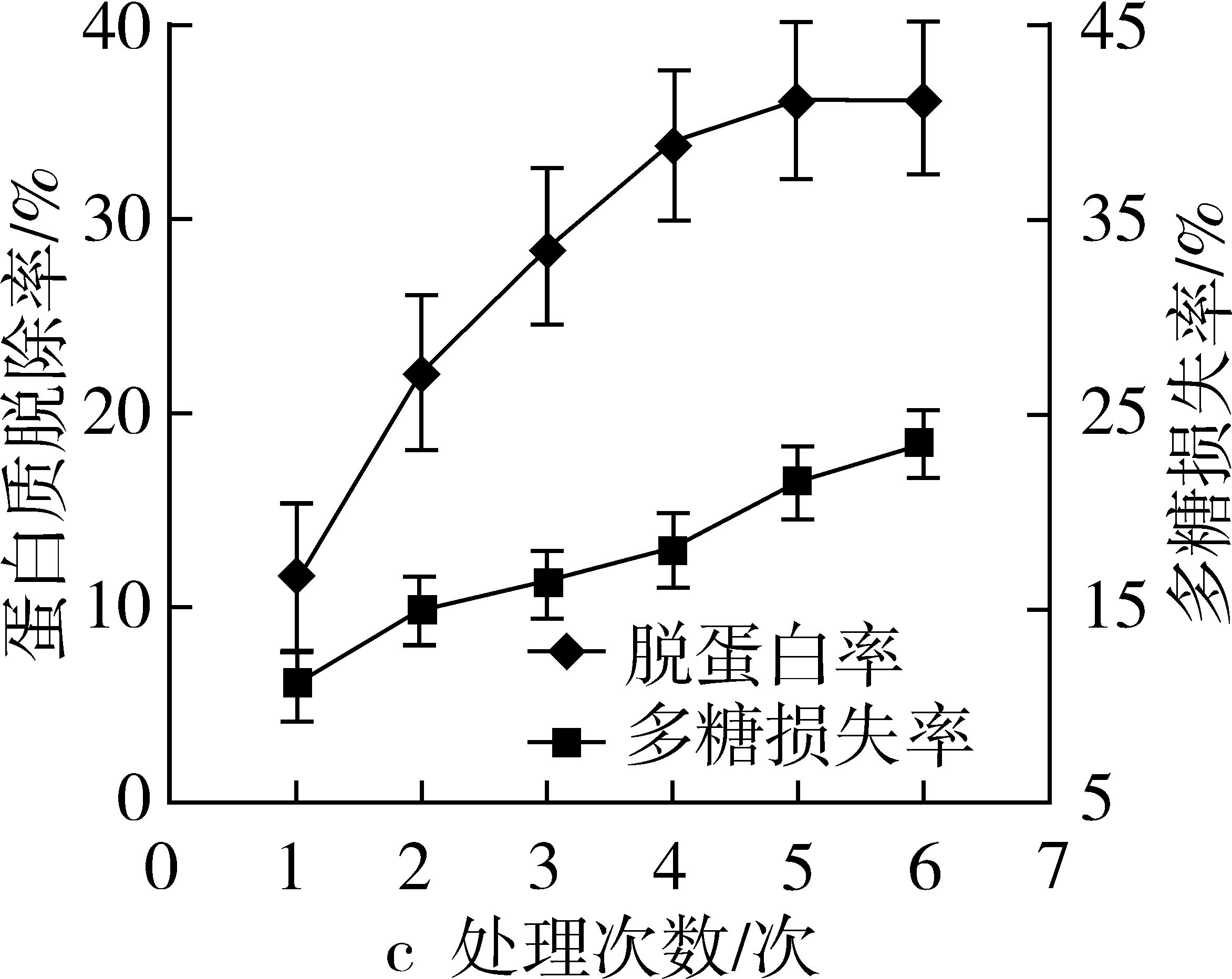
图3 亚铁氰化钾—乙酸锌法(a)、TCA法(b)和Sevag法(c)除蛋白的效果
注:样品经超声提取60 min;醇沉浓度为80%;苯酚硫酸显色法。
2.1.4 脱蛋白方法优化
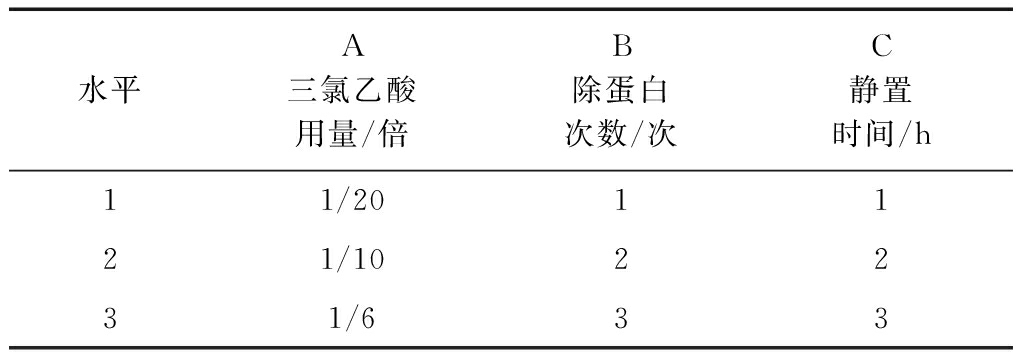
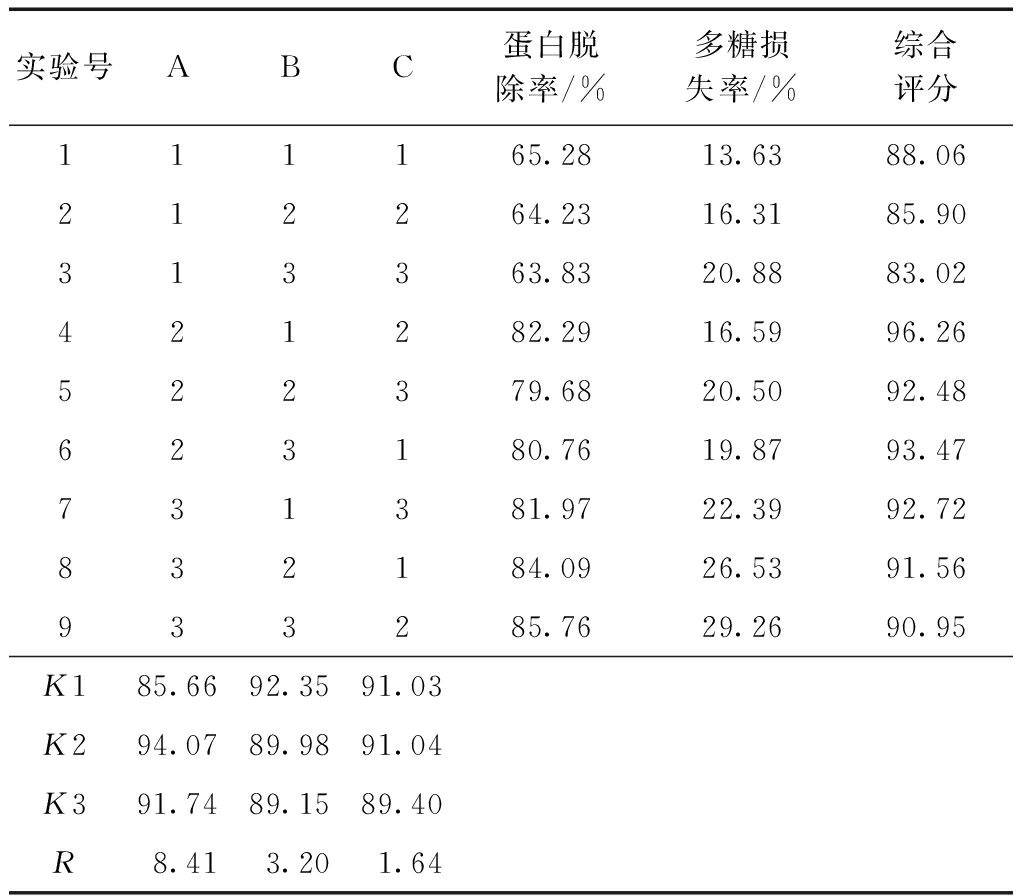
选取5%三氯乙酸的用量、除蛋白次数、静置时间为研究因素,每个因素选取3个水平,进行L9(33)正交实验,因素水平见表1。
表1 因素与水平
各取9份6 mL样液于具塞离心管中,按正交实验安排,分别加入质量分数为5%的三氯乙酸溶液进行除蛋白,离心后取上清,测其多糖和蛋白质含量。以蛋白脱除率和多糖损失率综合评分为指标,实验结果见表2,方差分析见表3。
表2 L9(33)正交实验设计结果
注:本实验采用综合加权评分法,权重系数均为0.5,分别把蛋白脱除率和多糖保留率最大的指标定为100分,各号按下式评分:综合评分=(蛋白质脱除率/85.76)×100×0.5+[(100-多糖损失率)/(100-13.63)]×100×0.5[11]。
由表2可知,各因素对脱蛋白效果的影响显著性大小顺序为:A>B>C,即三氯乙酸用量对脱蛋白效果影响最大,脱蛋白次数次之,静置时间对脱蛋白效果影响最小。根据各因素水平平均值K可得三氯乙酸法脱蛋白最佳工艺方案为A2B1C2,即以样液体积1/10倍量的5%三氯乙酸溶液,除蛋白1次,静置2 h为除蛋白的优选方法。
由方差分析结果(表3)可知,3个因素对脱蛋白效果影响显著性大小与极差分析相符合。其中,三氯乙酸用量对多糖脱蛋白效果具有极显著影响,脱蛋白次数和静置时间分别对脱蛋白效果有显著影响。
表3 方差分析
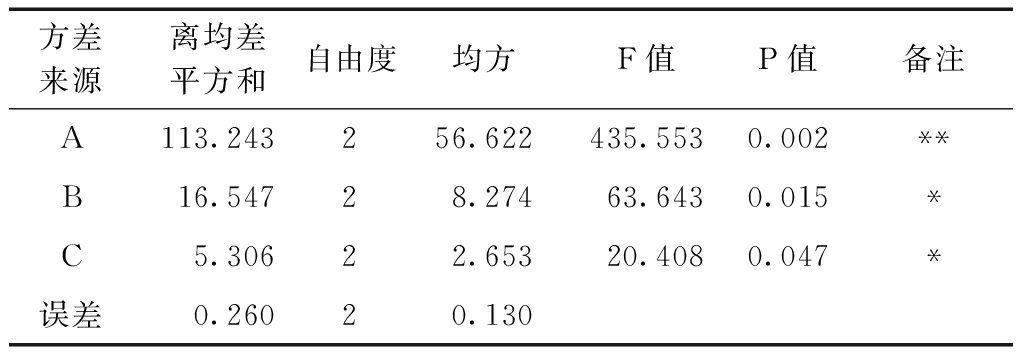
注:**表示差异极显著(P<0.01);*表示差异显著(P<0.05)。
2.1.5 显色方法
分别选用苯酚硫酸法和蒽酮硫酸法进行显色,比较不同显色方法下同一样品的测定结果的差异,结果见图4。
由图4可知,蒽酮硫酸法测定结果要低于苯酚硫酸法,且重现性、稳定性所测数据差异较大(RSD=5.86%)。这是由于蒽酮试剂稳定性较差,易氧化,遇水后测定数值波动较大,而苯酚硫酸法更易操作,其结果稳定性和重复性较好(RSD=3.24%),这与文献[12]报道相符。故显色方法采用苯酚硫酸法。
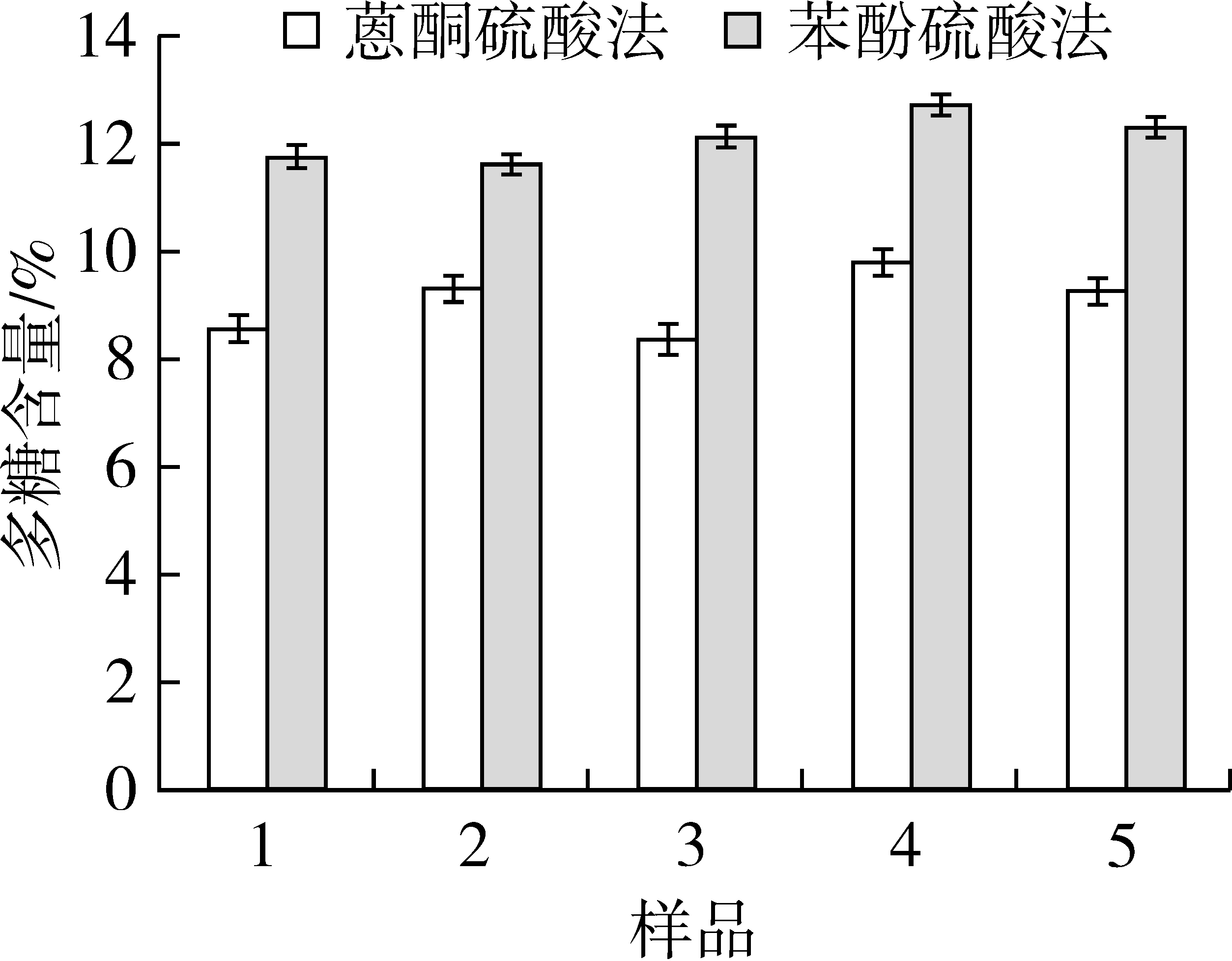
图4 不同显色方法对多糖含量测定的影响
注:样品采用超声提取60 min;醇沉终浓度为80%;加入样液体积1/10倍量、5%三氯乙酸溶液,静置2 h进行脱蛋白处理。
2.2 方法学验证
2.2.1 精密度实验
吸取同一供试品溶液,按苯酚硫酸法显色并测定其吸光度,平行测定六次,结果表明,相对标准偏差RSD=0.73%,小于5.0%,表明精密度良好。结果见表4。
表4 精密度测试结果(n=6)

2.2.2 稳定性实验
吸取同一供试品溶液,按苯酚硫酸法显色并测定其吸光度,每隔20 min测定1次,连续2 h,测得吸光度的RSD=0.91%,表明供试品溶液在2 h内显色稳定。结果见表5。
表5 稳定性实验结果(n=6)

2.2.3 重现性实验
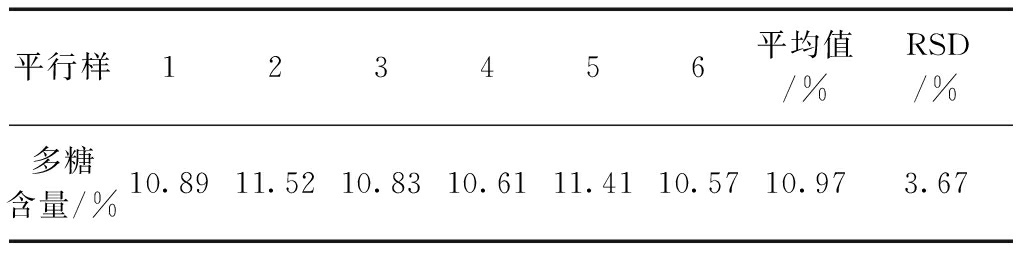
取同一批清平片样品6份,精密称定,按供试品溶液的制备方法制备,各取供试品溶液0.2 mL,按苯酚硫酸法测定吸光度,由回归方程计算供试品溶液中葡萄糖浓度,得出样品中多糖的含量,结果RSD=3.67%,表明该方法重现性良好。结果见表6。
表6 重现性实验结果(n=6)
2.2.4 加样回收实验
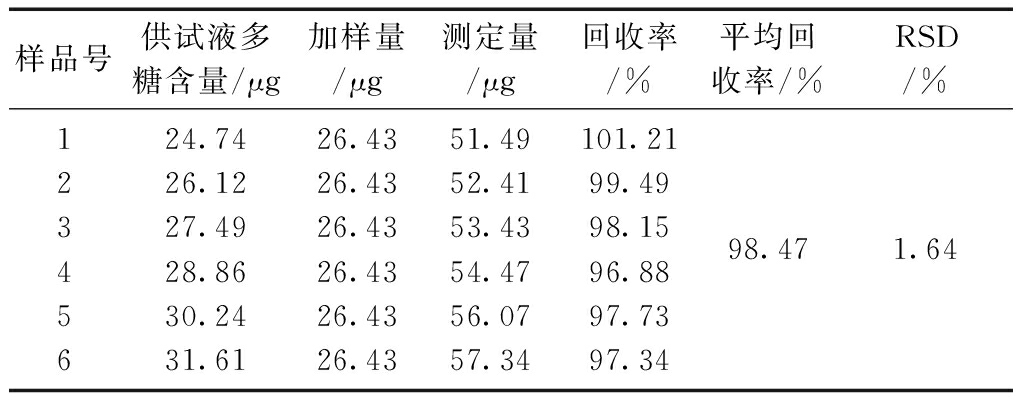
精密移取供试品溶液约0.1 mL于具塞刻度比色管中,加入标准葡萄糖溶液0.2 mL,补水至2.0 mL,平行制备6份,按苯酚硫酸法显色并测定其吸光度,计算回收率,结果平均加样回收率为98.47%,RSD为1.64%,表明该方法的准确度良好。结果见表7。
表7 回收率实验结果(n=6)
2.3 多糖含量测定
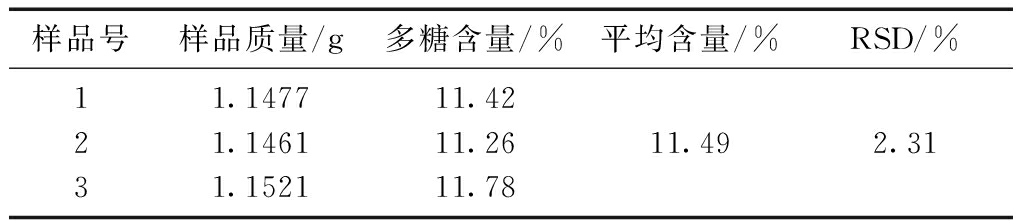
取同一批次的样品3份,精密称定,按供试品溶液的制备方法制备,各取供试品溶液0.2 mL,按苯酚硫酸法测定吸光度,由回归方程计算供试品溶液中葡萄糖浓度,得出样品中多糖的平均含量为11.49%,结果见表8。
表8 多糖含量测定结果
本研究对辅助降血糖保健食品中水溶性多糖含量测定的提取方式、醇沉浓度、脱蛋白方法、显色方法进行了单因素考察,同时对脱蛋白方法中沉淀剂的用量、除蛋白次数、静置时间进行了正交实验考察,优选出最佳样品前处理方法和显色方法,并对该方法进行了系统的方法学验证。通过实验,以稳定、简便可行的样品前处理方法消除小分子杂质和蛋白质等因素的干扰;以经典、稳定可靠的苯酚硫酸法为显色方法,对保证含多糖类的降血糖保健食品的质量具有重要的意义。
参考文献:
[1]于村, 丁钢强, 俞莎, 等. 香菇多糖测定的方法学研究[J]. 中国公共卫生, 2000, 16(3): 245-246.
[2]王慧玲, 张晓霞, 韩美艳, 等. 桔梗多糖的含量测定[J]. 郑州铁路职业技术学院学报, 2011, 23(3): 44-46.
[3]赵艳, 毕荣宇, 牟德华. 真菌多糖定量检测方法研究进展[J]. 食品与机械, 2012, 28(6): 264-267.
[4]郭志烨, 韩丽, 杨明, 等. 中药多糖定量测定方法的探讨[J]. 中成药, 2014, 36(10): 2172- 2176.
[5]王光亚. 保健食品功效成分检测方法[M]. 北京: 中国轻工业出版社, 2002: 19 -23.
[6]刘晓涵, 陈永刚, 林励, 等. 蒽酮硫酸法与苯酚-硫酸法测定枸杞子中多糖含量的比较[J]. 食品科技, 2009, 34(9): 270- 272.
[7]张惟杰. 糖复合物生化研究技术, 第二版[M]. 杭州: 浙江大学出版社, 1999: 11-13.
[8]魏苑, 张盛贵. 蒽酮- 硫酸法测定枸杞多糖含量的研究[J]. 食品工业科技, 2011, 32(3): 399- 401.
[9]李合生. 植物生理生化实验原理合和技术[M]. 北京: 高等教育出版社, 2000: 184-185.
[10]活泼, 蒋家新, 黄光荣. 香菇多糖提取工艺的研究[J]. 粮油食品科技, 2003, 11(3): 15-17.
[11]贾淑珍, 王成忠, 于功明. 香菇多糖脱色方法的研究[J]. 食品科技, 2007(6): 113-115.
[12]钟方晓, 任海华, 李岩. 多糖含量测定方法比较[J]. 时珍国医国药, 2007, 18(8): 1916-1917.●![]()
LIU Shi-juan, ZHANG Hai-tao, WANG Jing, XU Zhen-qiu
(Jiangsu Kanion Pharmaceutical Co., Ltd.,Lianyungang Jiangsu 222001)
Abstract:To establish a method for determination of the content of water- soluble polysaccharides in auxiliary hypoglycemic health food, the effects of extraction method, concentration of ethanol for precipitation, deproteinization method and coloration method on the determined values of polysaccharides content were studied. Result showed that the phenol- sulfuric acid method was chosen as the coloration method, with the conditions as follows: ultrasonic power 250 W, extraction time 60 min, 80% ethanol concentration for precipitation, and 1/10 polysaccharide extraction volume, 5% trichloroacetic acid concentration for deproteinization, deproteinization once, standing time 2 h, and the result was good. The standard curve regression equation wasA=0.0144C-0.0655, the calibration curve had a good linearity in the range of 6.61-79.28 μg/mL for glucose with the correlation coefficient (r) of 0.9996, and the average recovery at the spiked concentrations was 98.47% with RSD of 1.64%. The average content of water- soluble polysaccharides from the auxiliary hypoglycemic health food was 11.49% with RSD of 2.31%. This method was simple and accurate with a good repetion, which can be used to determine the content of polysaccharides in auxiliary hypoglycemic health products.
Key words:water-soluble polysaccharides; determination method; phenol- sulfuric acid method; health food
收稿日期:2015-07-28
作者简介:刘世娟,1971年出生,女,硕士.
中图分类号:TS 201.4
文献标志码:A
文章编号:1007-7561(2016)01-0050-05